https://jamanetwork.com/journals/jama/fullarticle/2768926
Question Are genetic variants associated with severe coronavirus disease 2019 (COVID-19) in young male patients?
Findings In a case series that included 4 young male patients with severe COVID-19 from 2 families, rare loss-of-function variants of the X-chromosomal TLR7 were identified, with immunological defects in type I and II interferon production.
Meaning These findings provide insights into the pathogenesis of COVID-19.
Importance Severe coronavirus disease 2019 (COVID-19) can occur in younger, predominantly male, patients without preexisting medical conditions. Some individuals may have primary immunodeficiencies that predispose to severe infections caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
Objective To explore the presence of genetic variants associated with primary immunodeficiencies among young patients with COVID-19.
Design, Setting, and Participants Case series of pairs of brothers without medical history meeting the selection criteria of young (age <35 years) brother pairs admitted to the intensive care unit (ICU) due to severe COVID-19. Four men from 2 unrelated families were admitted to the ICUs of 4 hospitals in the Netherlands between March 23 and April 12, 2020. The final date of follow-up was May 16, 2020. Available family members were included for genetic variant segregation analysis and as controls for functional experiments.
Exposure Severe COVID-19.
Main Outcome and Measures Results of rapid clinical whole-exome sequencing, performed to identify a potential monogenic cause. Subsequently, basic genetic and immunological tests were performed in primary immune cells isolated from the patients and family members to characterize any immune defects.
Results The 4 male patients had a mean age of 26 years (range, 21-32), with no history of major chronic disease. They were previously well before developing respiratory insufficiency due to severe COVID-19, requiring mechanical ventilation in the ICU. The mean duration of ventilatory support was 10 days (range, 9-11); the mean duration of ICU stay was 13 days (range, 10-16). One patient died. Rapid clinical whole-exome sequencing of the patients and segregation in available family members identified loss-of-function variants of the X-chromosomal TLR7. In members of family 1, a maternally inherited 4-nucleotide deletion was identified (c.2129_2132del; p.[Gln710Argfs*18]); the affected members of family 2 carried a missense variant (c.2383G>T; p.[Val795Phe]). In primary peripheral blood mononuclear cells from the patients, downstream type I interferon (IFN) signaling was transcriptionally downregulated, as measured by significantly decreased mRNA expression of IRF7, IFNB1, and ISG15 on stimulation with the TLR7 agonist imiquimod as compared with family members and controls. The production of IFN-γ, a type II IFN, was decreased in patients in response to stimulation with imiquimod.
Conclusions and Relevance In this case series of 4 young male patients with severe COVID-19, rare putative loss-of-function variants of X-chromosomal TLR7 were identified that were associated with impaired type I and II IFN responses. These preliminary findings provide insights into the pathogenesis of COVID-19.
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has resulted in a pandemic with more than 9 million infections of coronavirus disease 2019 (COVID-19) since the first reported cases in December 2019. SARS-CoV-2 is the most recently identified member of the Betacoronaviruses and is transmitted through highly contagious respiratory droplets.1 The clinical spectrum of COVID-19 ranges from asymptomatic disease or mild symptoms of the upper respiratory tract in most cases to potentially lethal, severe pneumonia with acute respiratory distress syndrome (ARDS) in up to 5% of patients whose test results were positive for COVID-19.2,3 Cohort studies have identified older age, male sex, and comorbidities such as hypertension, diabetes, and obesity as risk factors predisposing to severe disease.2-5 It is unclear to what extent specific genetic factors may explain the predisposition of individuals to develop severe COVID-19 requiring admission to the intensive care unit (ICU).
Severe COVID-19 is unusual in younger patients without preexisting medical conditions. In the Netherlands, data from May 14, 2020, published by the National Institute for Public Health (RIVM) (eFigure 1 in the Supplement) showed that 3.5% (399/11 430) of hospitalized patients with COVID-19 were younger than age 35 years.6 For this age group, 6 of the 7 patients who died were men. Despite these small numbers, a male-biased excess in case-fatality rates has also been described in various studies, which included patients younger than 35 years.7-9
In severely affected young men, and in particular in brother pairs (sharing half of their genomes, with an increased chance of identifying an X-linked disease), a unique genetic defect might be present that could indicate a genetic predisposition to contract coronavirus infections. The purpose of this case series was to explore whether such genetic variants could be identified in patients with severe COVID-19.
This case series was conducted at the Radboud University Medical Center (RUMC), Nijmegen, the Netherlands, which was the coordinating center for COVID-19 research in the regional network. The only 2 male brother pairs younger than 35 years with SARS-CoV-2 infection confirmed by real-time reverse transcriptase–polymerase chain reaction (PCR) assay of nasal and pharyngeal swabs, admitted to the ICU of the RUMC or one of the ICUs in the network between March 23 and April 25, 2020, were enrolled. The stringent selection of young, severely affected brother pairs was chosen to increase the chance of identifying monogenic, possibly X-linked, factors. Family members were included for segregation analysis of genetic variants and as controls in functional experiments.
Patients and relatives provided written consent for diagnostic whole-exome sequencing, which entailed complete exome analysis according to hospital procedures and was arranged under ethical approval obtained from the regional Arnhem and Nijmegen Ethics Committee. Explicit written consent was given for publication of research findings.
For index cases and parents of family 1, genomic DNA was isolated from 4-mL whole EDTA blood using an automated standard procedure. Genomic DNA of patient II-2 from family 1 was isolated from stored sputum samples, while genomic DNA of patient II-2 from family 2 was derived from buccal swabs.
Rapid whole-exome sequencing was performed in both families, similar to previous reports with some modifications.10 In brief, DNA samples were processed using the Human Core Exome Kit and extended RefSeq targets (Twist Biosciences). Libraries were prepared according to the manufacturers’ protocols. All DNA samples were sheared using a Covaris R230 ultrasonicator (Covaris), subsequently followed by 2 × 150–base pair paired-end sequencing on a Novaseq 6000 instrument (Illumina).
Mean sequence coverage was 116-fold (II-1 from family 1) and 103-fold (II-1 from family 2), respectively, and 98.8% and 99.0% of all target bases read 20-fold or greater, sufficient for reliable variant calling (for details, see eTable 1 in the Supplement). Downstream processing was performed using an automated data analysis pipeline that included Burrows-Wheeler Aligner mapping, Genome Analysis Toolkit variant calling, and custom-made annotation.11 For trio-based analysis (family 1), libraries were sequenced simultaneously to favor interpretation of results; exome analysis of the affected brothers (II-2 from family 1 and II-2 from family 2) was also performed to check segregation of all rare filtered variants in the respective index patients.
A diagnostic in-silico gene-panel analysis (version DG2.18) was conducted similar to previous reports, now encompassing a list of 420 established primary immunodeficiency genes, and turned out negative.12,13 Subsequently, an exome-wide research-based reanalysis was applied using standard filtering steps for (1) rare nonsynonymous and (2) possibly damaging variants that occur in genes with previously described immune function (eTable 2 in the Supplement). Initially, analysis and filtering of variants in both index patients was performed separately, taking into account the segregating variants in their respective affected brothers, with the exclusion of paternally inherited variants in family 1, to narrow down the list of candidate genes (eTable 3 in the Supplement). Additionally, an overlap strategy14 was applied between the segregating rare variants of all affected brothers to identify possible overlapping genes with rare variants between the families.
All identified TLR7 variants were confirmed by standard Sanger sequencing and co-segregation analysis was consecutively performed in all available family members (eFigure 2, primer sequences are given in eTable 4 in the Supplement).
Venous blood was drawn and collected in EDTA tubes (Monject). Subsequently, peripheral mononuclear blood cell (PBMC) isolation was conducted as described elsewhere.15 In brief, the PBMC fraction was obtained by density centrifugation of blood, diluted 1:1 in pyrogen-free saline over Ficoll-Paque (Pharmacia Biotech). Cells were washed twice in saline and suspended in cell culture medium (Roswell Park Memorial Institute [RPMI] 1640, Gibco) supplemented with gentamicin, 10 mg/mL; L-glutamine, 10 mM; and pyruvate, 10 mM. PBMC stimulations were performed with 5 × 105 cells/well in round-bottom 96-wells plates (Greiner) for either 4 hours (for transcription of type I interferon [IFN] genes) and 7 days (for cytokine production) in the presence of 10% human pool serum at 37 °C and 5% carbon dioxide. Next to a negative RPMI control, the toll-like receptor 7 (TLR7) agonist imiquimod (imidazoquinoline compound, Invivogen) was used in varying concentrations (2.5 μg/mL and 5 μg/mL) and heat-killed Candida albicans yeast (1 × 106/mL). After the incubation period and a centrifugation step, supernatants were collected and stored at −20 °C until measured using enzyme-linked immunosorbent assay. The remaining cell pellets were resuspended in RLT buffer (Qiagen) and snap frozen to be stored at −80 °C until processing for RNA isolation.
Quantitative PCR (qPCR) assays were performed following standard methods to assess expression levels of TLR7 (OMIM 300365, eTable 5 in the Supplement). To evaluate the host type I IFN response in patient PBMCs, the downstream mRNA expression levels were measured for type I IFN genes (IRF3, OMIM 603734; IRF7, OMIM 605047; ISG15, OMIM 147571; IFNB1, OMIM 147640) and all were compared with a housekeeping gene (HPRT1, OMIM 145000). First, to explore the pathogenicity of the mutations, PBMCs were collected and purified. Cells were stimulated for 4 hours in vitro in RPMI cell culture medium with or without the TLR7 agonist imiquimod, which is known to induce TLR7 expression.16 RNA was isolated from PBMCs using the RNeasy Mini kit (Qiagen) following the manufacturer’s instructions. All obtained material (~ 350 ng) was used for subsequent cDNA synthesis using iScript cDNA Synthesis Kit (Bio-Rad). The experiment was performed with 3 technical replicates using GoTaq qPCR master mix (Promega) and 7 primers sets (summarized in eTable 6 in the Supplement). PBMCs of patient II-2 from family 2 were processed at a later stage for a separate qPCR experiment, the results of which have been included in eFigure 3 in the Supplement.
The cytokines were measured following the instructions of the manufacturer. Supernatants after the 7-day stimulation were measured for IFN-γ (R&D Systems and PeliKine Compact, Sanquin). The IFN-γ response was assessed due to its implicated role in TLR7-dependent antiviral host defense, especially for single-stranded RNA (ssRNA) viruses.17 Therefore, the capacity of PBMCs to induce an IFN-γ–specific response to the TLR7 ligand imiquimod was tested. The IFN-γ response to a heterologous stimulus unrelated to the TLR7 pathway, the fungal pathogen C albicans, was measured as a positive control.
Descriptive statistics were used for patient characteristics. Continuous variables were described using mean values with range intervals. For the statistical simulations to estimate the probability of identifying the same gene harboring rare variants in 2 index patients from 2 independent families, the a priori chance was calculated using exome sequencing data of 4776 healthy parents of patients with intellectual disability (median 85 × coverage, Agilent SureSelect v4 and Surelect v5).18 For each exome, variants were selected according to the same filtering steps as outlined in eTable 2 in the Supplement in genes that are associated with an annotated function in the immune system (Gene Ontology term contains “immune”/“immuno” or there is a mouse knockout phenotype associated with the immune system [based on Mouse Genome Informatics]). These variants were then expressed in matrix D = {dij}n × m, where dij∈{0, 1}, n is the number of individuals (n = 4776), m is the number of genes with at least 1 variant in any individual (m = 2271), and dij is 1 if the individual had 1 or more variants in this gene and 0 otherwise. Given this matrix, a new matrix C = DDT could be calculated of size n × n, where each element of the new matrix cij equals the number of genes with at least 1 variant in common between individual i and individual j. Of note, this matrix C = {cij}n × n is symmetric. A new matrix P = {pij}n × n was then constructed, where
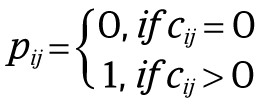
This matrix P is also symmetric. In addition, the following quantities were introduced: v = ∑ i > jpij is the number of pairings in which 2 individuals i and j share a variant in the same gene, and k = n(n −1)/2 is the number of total pairings. Given these quantities, the probability of 2 individuals sharing a variant in the same gene can be calculated as P1 = v/k.
The probability of the identification of rare, loss-of-function, or highly conserved missense variants in the gene TLR7 in 2 unrelated individuals was determined. For this purpose, all loss-of-function and missense variants in TLR7 were extracted from the exomes in the gnomAD 2.1.0 database according to the same filtering steps as outlined in eTable 2 in the Supplement. It was assumed that the individuals in this database are a reasonable genetic representation of the normal population. From the extracted variants, the variant allele frequency was summed to estimate the proportion of individuals carrying at least 1 such variant in the TLR7 gene, assuming a worst-case scenario in which each individual in gnomAD carries at most 1 such variant. By using the allele frequency as calculated by gnomAD, the different number of alleles for males and females were taken into account, as well as alleles that cannot be assessed due to, for example, lack of sequence coverage.
For the qPCR experiments, the fold change mRNA levels per gene derived from the averaged triplicate values were compared between the individual patients and pooled controls from the same experiment by means of a 2-sided t test on the log-transformed fold changes. P values (unadjusted) below .05 were considered statistically significant. Missing values in the qPCR experiments arising from technical error were excluded from analysis. A single value was removed from analysis because it was considered to be an outlier; it was calculated that this value would be 42 × deviant from the mean value based on the highest standard deviation of this sample for all other genes (eTable 5 in the Supplement). These analyses were conducted in R version 3.6.1 (R Project for Statistical Computing). In case of missing data in patient characteristics, the respective variables were excluded from the analysis.
Patient II-1 from family 1, index patient of family 1, was a 32-year-old man of Dutch ancestry (Figure 1A) who presented with dyspnea and hypoxia with diffuse bilateral ground-glass opacities of the lungs on computed tomographic scan due to proven COVID-19. His medical history was unremarkable apart from a chronic nonallergic rhinosinusitis (Table). On the day of hospital admission, the patient was transported to the ICU for noninvasive oxygen supplementation. Because of progressive respiratory insufficiency 2 days after admission, the patient was intubated and mechanically ventilated. After 11 days in the ICU, he was successfully extubated after significant respiratory improvement. He was discharged from the ICU to the clinical ward at day 16 with minimal supplementation of oxygen. However, at day 20, the patient experienced dyspnea and thoracic pain, which appeared to be caused by a left-sided pneumothorax, secondary to giant bullae that developed during or after his stay in the ICU. A chest drain was inserted, and the patient was transferred to the RUMC for continuation of treatment and the possibility of surgical intervention. The patient’s condition remained stable over the course of his stay at the ward of the pulmonology department, and subsequent radiographical follow-up showed a slow but steady improvement of the pneumothorax. After 9 days, the drain was removed, and the patient was discharged from hospital to continue medical rehabilitation at home. His 29-year-old, previously healthy brother, patient II-2 from family 1, also contracted infection with SARS-CoV-2, despite living in a separate household. He was admitted to the ICU and exhibited spiking fevers with shock, as evidenced by hypotension refractory to treatment with inotropic agents and corticosteroids, caused by severe COVID-19 with a concurrent secondary bacterial infection, and did not survive. Both parents, of whom the father tested positive for SARS-CoV-2, had mild to moderate respiratory symptoms (Table; Figure 1A; eTable 7 in the Supplement).
Patient II-1 from family 2, index patient of family 2, was a 21-year-old man of African ancestry (Figure 1A) with an unremarkable medical history (Table). He was admitted in hospital with proven COVID-19. A computed tomographic scan of the lungs showed bilateral ground-glass opacities and consolidations and additional subtle subsegmental pulmonary embolisms, for which anticoagulants were started. Because of imminent respiratory insufficiency, the patient was initially treated with high-flow oxygen via a nasal cannula. Subsequently, due to respiratory muscle exhaustion, the patient was intubated and mechanically ventilated in the ICU. The patient was transferred to the RUMC. After 11 days of respiratory improvement, the patient was successfully extubated and later discharged to the clinical ward for further medical rehabilitation. His brother, patient II-2 from family 2, 23 years of age and living in the same household, was also admitted to the ICU with severe COVID-19. He was intubated and mechanically ventilated for a total of 9 days and was discharged to the clinical ward after extubation. Additional information on the clinical characteristics of all patients can also be found in the supplemental information (Table; eTable 7 in the Supplement).
Both separate and overlap analysis strategies independently identified X-chromosomal TLR7 as the most biologically plausible candidate gene in both families (eTable 2 and eFigure 3 in the Supplement), with literature on TLR7 supporting a direct function in the innate immune response against Coronaviruses.19-21 In patient II-1 from family 1, a 4-nucleotide hemizygous deletion was identified, leading to a predicted total loss of function of TLR7 (NM_016562.3: c.2129_2132del; p.[Gln710Argfs*18]) (Figure 1B). The deletion was also detected in the deceased brother, II-2 from family 1, and appeared to be heterozygous in the carrier mother, I-2 from family 1 (Figure 1; eFigure 2A in the Supplement). In patient II-1 from family 2, a missense variant (NM_016562.3: c.2383G>T; p.[Val795Phe]) was identified (Figure 1B), which was predicted as deleterious in all in-silico prediction tools (eAppendix in the Supplement). The resulting amino acid change affects the leucine-rich repeat region–carboxy-terminal domain, which is highly intolerant to variation (eFigure 4 in the Supplement). Segregation analysis confirmed the presence of the same missense amino acid change in the affected brother (eFigure 2B in the Supplement).
The a priori chance of finding an overlapping gene in 4 individuals from 2 families was simulated following the strategy outlined in the Methods section. The analysis of control exomes resulted in an average of 8.31 filtered variants per individual. In total, k = 11 402 700 pairings were simulated, resulting in v = 1 624 432 pairings with 2 variants in the same gene. Given these quantities, the probability was calculated of 2 individuals sharing a variant in the same gene as P1 = v/k = .14. Knowing that for these variants there was complete segregation of the X chromosome among the 2 brothers in each of the 2 families, a second probability can be calculated as P2 = .5 × .5 = .25. Because P1 and P2 are independent of each other, these events can be combined into an overall probability as P = .14 × .25 ≈ .04. This calculation did not consider a selection of the genes that are loss-of-function intolerant (pLI ≥ 0.9, n = 3230 of all genes).22
The probability of the identification of rare, loss-of-function, or highly conserved missense variants in the gene TLR7 in 2 unrelated individuals was determined as described in the Methods section. The allele frequencies of 94 unique loss-of-function and missense variants were extracted from population exome data. Based on the sum of allele frequencies, the probability of encountering an individual in gnomAD with such a variant is P1 = 1.078e−3. Therefore, the probability of encountering 2 such individuals is (P1)2 = 1.16e−4 ≈ P < .001.
TLR7 mRNA in index patient II-1 from family 1, carrying the TLR7 nonsense variant, could not be amplified in whole-blood (eFigure 5 in the Supplement). After stimulation with imiquimod, TLR7 expression was upregulated in PBMCs isolated from healthy controls, while all 3 tested patients from families 1 and 2 did not show enhanced expression of TLR7 (Figure 2A; eFigure 6A in the Supplement).
The 2 rare variants in TLR7 identified in the patients both resulted in defective upregulation of type I IFN–related genes in the TLR7 pathway (Figure 3) in response to the TLR7 agonist imiquimod as compared with controls, showing a statistically significant change (Figure 2B; eFigure 6B in the Supplement). More specifically, exposure to imiquimod resulted in increased mRNA expression of the genes IFNB1, IRF7, and ISG15 in PBMCs isolated from controls and the parents of index patient II-1 from family 1. In contrast, expression of downstream genes was defective in the 2 index patients and in the affected brother II-2 from family 2 (Figure 2B; eFigure 6B in the Supplement). Transcription of IRF3 was not induced in patients nor controls (Figure 2B).
In addition, both index patients were completely deficient for IFN-γ production in response to TLR7 stimulation (Figure 2C). In contrast, this response was intact on stimulation with C albicans, hence showing a normal capacity of patient PBMCs to produce IFN-γ (eFigure 7 in the Supplement).
In this case series of 4 young men from 2 unrelated families with severe COVID-19, unique loss-of-function variants in X-chromosomal TLR7 were identified. All patients required mechanical ventilation in the ICU, and 1 patient died. In PBMCs isolated from patients, the identified nonsense and missense TLR7 variants impaired mRNA expression of TLR7 on stimulation with the TLR7 agonist imiquimod, showing a loss-of-function effect. Furthermore, in these analyses, the patients displayed an impaired transcriptional host type I IFN response downstream of the TLR7 pathway, as evidenced by impaired upregulation expression of IRF7, IFNB1, and ISG15. Additionally, an abrogated production of the type II IFN, IFN-γ, was observed in the patients’ PBMCs stimulated with imiquimod, while the response to the heterologous stimulus C albicans was intact. Collectively, these data suggest an association between the presence of rare loss-of-function TLR7 variants in patients with severe COVID-19 and functional immunological defects of the type I and II IFNs.
To our knowledge, the TLR7 variants identified in this case series have not been described in the gnomAD browser nor in any other population database.23 In general, there is little variation in the TLR7 gene in population databases, which suggests that there is particular genetic variant constraint against predicted loss of function (pLI score, 0.98). Independent studies have also shown that TLR7, similar to other endosomal TLRs (TLR3, TLR8, and TLR9), is subject to evolutionary, selective constraints.24-26 The observed patterns of purifying selection and the constraint against loss of function of TLR7 in humans indirectly support a causation between the 2 unique loss-of-function variants and the immunological defects.25,26 To our knowledge, no patients have been described with recurrent infections due to a TLR7 deficiency, although a viral ligand—particularly an ssRNA virus—has long been suspected. The question has been raised whether the viruses responsible for the patterns of purifying selection observed for TLR7, TLR8, and TLR9 still exist in humans.25 The severe clinical course of COVID-19 in the young men described here suggests that SARS-CoV-2 is a virus that could be capable of exerting purifying selection on TLR7, as has been observed. The highly specific defect in host defense against SARS-CoV-2 and potentially other coronaviruses in these individuals with TLR7 deficiency has similarities with the specific susceptibility to herpes encephalitis in patients with defects in the TLR3 recognition pathway.27,28
This study identified the TLR7 pathway as an inducer of type I and II IFN responses in COVID-19. TLR7 has been implicated as an important pattern recognition receptor in the recognition of ssRNA of the Middle East respiratory syndrome CoV (MERS-CoV) and severe acute respiratory syndrome CoV (SARS-CoV) in murine infection models, making it a likely candidate to function as a central pattern recognition receptor in SARS-CoV-2.21 The importance of intact TLR7 signaling in COVID-19 is indirectly supported by research in murine infection models with MERS-CoV showing that this pathway is instrumental in the production of type I IFNs in airway epithelial cells.21 Moreover, whole-genome sequencing of SARS-CoV, MERS-CoV, and SARS-CoV-2 has demonstrated that the SARS-CoV-2 genome contains more ssRNA motifs that could interact with TLR7 than the SARS-CoV genome, indicating that TLR7 signaling might be even more relevant in the pathogenesis of COVID-19.20 Coronaviruses have various mechanisms to evade the innate immune response, especially by modifying the type I IFN response.29 In comparison with other respiratory viruses, SARS-CoV-2 induces a lower antiviral transcriptional response marked by low type I IFN levels and elevated chemokine expression.30 Furthermore, patients with severe COVID-19 exhibit an impaired type I IFN response and a lower viral clearance.31,32 It can be hypothesized that TLR7 deficiency leads to impaired viral clearance with a high viral load, thereby increasing the direct cytopathic viral effects and ensuing hyperinflammatory response, which puts these individuals at risk for severe COVID-19. However, no clinical data on viral titers were available to assess this hypothesis.
This was a case study of young patients with COVID-19 and severe clinical disease, but it is possible that some individuals with TLR7 deficiency exhibit milder disease. Given the low prevalence of loss-of-function alleles of TLR7 in the general population, TLR7 deficiency is likely to be very rare. Other differences in susceptibility to COVID-19 may have a more complex multigenic or multifactorial basis; large population-based studies would be needed to demonstrate association with the underlying genetic variation. Recently, a first association was established for a 3p21.31 gene cluster as a genetic susceptibility locus in patients with COVID-19 and respiratory failure.33 Thus far, no evidence for common low effect size variants in TLR7 has been described, although such variants might affect immune function, as shown for other immune-related disorders.34-37 In recent literature, more common genetic variation in TLR7 has been proposed as a possible explanation of the male sex bias in COVID-19 because of its localization on the X chromosome and well-established function in innate immunity.7 TLR7 escapes X inactivation, which could explain the higher basal expression levels of TLR7 and more prominent TLR7-induced type I IFN responses that have been observed in women compared with men.34,38 Conversely, the elevated TLR7 expression in women has been linked to an increased susceptibility to develop autoimmune disease, including systemic lupus erythematosus, that could be related to an enhanced type I IFN response.39 It is possible, but only speculative, that the differences in TLR7 dosage between men and women could in part explain the predisposition of men to develop severe COVID-19. The TLR7 dosage could range depending on the number of functional gene copies, from (1) the immunodeficient patients presented here as the most severe outliers with no intact TLR7 copy, (2) healthy men with 1 intact TLR7 copy, and (3) healthy women with 2 active TLR7 copies. However, it is unknown whether this gene dosage effect could be affected by common genetic variation in TLR7, though the most recent work suggests such a role for common TLR7 variants.40
This study has several limitations. First, the case series precludes drawing conclusions regarding causality between the rare loss-of-function TLR7 variants and the pathogenesis of severe COVID-19. However, the likelihood calculations presented in this study add statistical credibility to rule out that the identification of 2 individuals with shared variation in TLR7 is explained by chance.
Second, the functional experiments with IFN-γ measurements lacked statistical significance, possibly due to the limited number of replications and controls included in this study. However, these measurements were below the assay detection limit only in the patients, compared with the familial controls, who exhibited normal responses to C albicans in the same experiment.
In this case series of 4 young male patients with severe COVID-19, rare putative loss-of-function variants of X-chromosomal TLR7 were identified that were associated with impaired type I and II IFN responses. These preliminary findings provide insights into the pathogenesis of COVID-19.
Accepted for Publication: July 10, 2020.
Published Online: July 24, 2020. doi:10.1001/jama.2020.13719
Concept and design: van der Made, Simons, Schuurs-Hoeijmakers, van der Schoot, J. van der Meer, Brunner, Netea, van de Veerdonk, Hoischen.
Acquisition, analysis, or interpretation of data: van der Made, Simons, Schuurs-Hoeijmakers, van den Heuvel, Mantere, Kersten, van Deuren, Steehouwer, van Reijmersdal, Jaeger, Hofste, Astuti, Corominas Galbany, van der Hoeven, Hagmolen of ten Have, Klijn, C. van der Meer, Fiddelaers, de Mast, Bleeker-Rovers, Joosten, Yntema, J. van der Meer, Gilissen, Nelen, Brunner, van de Veerdonk, Hoischen.
Drafting of the manuscript: van der Made, Simons, Schuurs-Hoeijmakers, van Deuren, Steehouwer, van Reijmersdal, Hofste, Astuti, Corominas Galbany, van der Schoot, van der Hoeven, Hagmolen of ten Have, Klijn, C. van der Meer, Fiddelaers, Bleeker-Rovers, Yntema, Nelen, Brunner, van de Veerdonk, Hoischen.
Critical revision of the manuscript for important intellectual content: van der Made, Simons, Schuurs-Hoeijmakers, van den Heuvel, Mantere, Kersten, Jaeger, van der Hoeven, C. van der Meer, de Mast, Joosten, J. van der Meer, Gilissen, Brunner, Netea, van de Veerdonk, Hoischen.
Statistical analysis: van der Made, van Deuren, Gilissen.
Obtained funding: Hoischen.
Administrative, technical, or material support: Simons, Schuurs-Hoeijmakers, van den Heuvel, Kersten, Steehouwer, van Reijmersdal, Jaeger, Hofste, Astuti, Corominas Galbany, van der Schoot, Hagmolen of ten Have, Klijn, C. van der Meer, Fiddelaers, J. van der Meer, Nelen.
Supervision: Schuurs-Hoeijmakers, de Mast, Yntema, J. van der Meer, Netea, van de Veerdonk, Hoischen.
Conflict of Interest Disclosures: Dr Joosten reported being a scientific founder of Trained Therapeutic Discovery and a scientific advisory board member of Olatec Therapeutics. Dr Netea reported being a scientific founder of Trained Therapeutic Discovery and receiving grants from ViiV HealthCare outside the submitted work. No other disclosures were reported.
Funding/Support: Ms Steehouwer and Drs Gillissen, Brunner, and Hoischen are supported by the Solve-RD project. The Solve-RD project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No. 779257. Dr Netea was supported by an European Research Council Advanced Grant (No. 833247) and a Spinoza Grant of the Netherlands Organization for Scientific Research. Dr van de Veerdonk was supported by a ZonMW (The Netherlands Organization for Health Research and Development) Vidi grant (No. 91718351). Dr Mantere was supported by the Sigrid Jusélius Foundation. This research was part of the Netherlands X-omics Initiative and partially funded by NWO (The Netherlands Organization for Scientific Research; project 184.034.019) and Radboud Institute for Molecular Life Sciences PhD grants (to Drs Hoischen and Netea).
Role of the Funder/Sponsor: The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Additional Contributions: We thank the patients and their families for their participation. We thank Radboud University Medical Center (RUMC) personnel: W. Melchers, PhD; S. D. van der Velde-Visser; M. M. H. M. Jacobs-Camps, M. van de Vorst, BSc; G. Khazeeva, MSc; all colleagues of the multidisciplinary immuno-exome sign-out meeting (MDO); all colleagues of the diagnostic primary immunodeficiency–exome interpretation group, in particular: J. H. S. Diepstra, BSc, E. P. D. Hoenselaar, BSc, and M. Weiss, PhD; the COVID-19 clinical team, in particular: W. Hoefsloot, MD, PhD, J. Hoogerwerf, MD, PhD, M. Reijers, MD, PhD; the Radboud University Medical Center Center for Infectious Diseases (RCI) COVID-19 team; and all colleagues at the Laboratory for Experimental Internal Medicine, in particular H. Lemmers, BSc, F. Weren, MSc, H. Dijkstra, BSc, L. van Emst, BSc, and L. Fransman, BSc. We also thank R. A. Willemze, MD, PhD, Viroscience, Erasmus MC, Rotterdam, the Netherlands. None of the persons indicated above received compensation for their role in the study.



Nessun commento:
Posta un commento